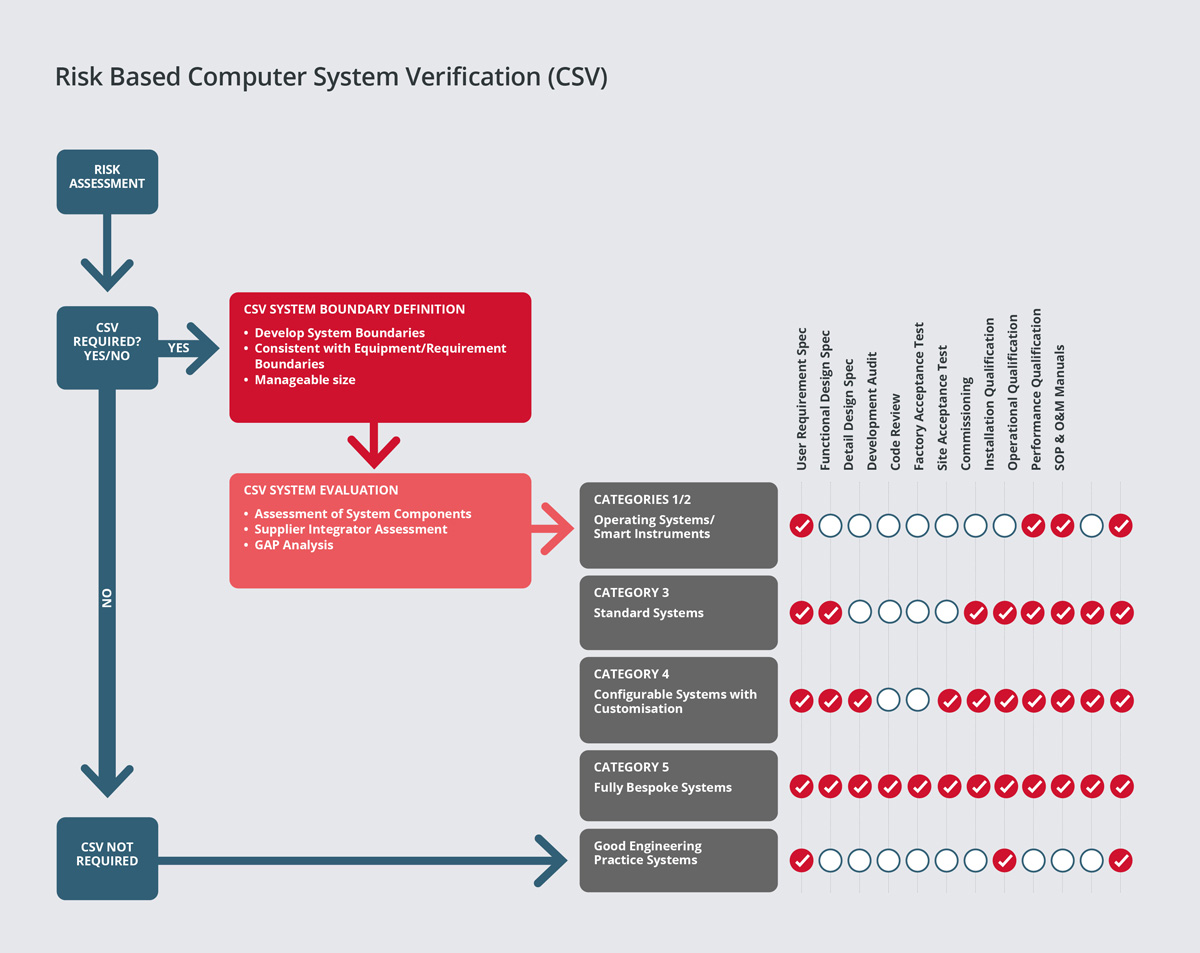
Our consultants have extensive experience of conducting risk assessments and criticality analysis for process facilities. Our team would typically carryout risk assessments and criticality analysis during the design phase of the project, in order to classify systems into ‘critical’ and ‘non-critical’ impact systems. This is significant, as critical systems or high-risk systems will require a higher level of documentation, field inspections and testing.
User Requirement Specifications (URS) are critical documents as their primary purpose is to document, at a high level, what users need from facilities, systems or utilities. They are also the basis for design, procurement, commissioning, qualification & validation activities.
Honed in the highly demanding world of regulated industries and products, our engineers and scientist have considerable experience in supporting our customers with the preparation of URS’s for a variety of industries.
We have experience of many different technologies and have prepared and executed protocols for Design Qualification (DQ), Installation Qualification (IQ), Operational Qualification (OQ), Performance Qualification (PQ), Cleaning Validation, Process Qualification and Analytical Method Validation for many of these technologies. We have an extensive library of protocols, which can be tailored to your needs. For commonly used equipment we have protocols which can be quickly retrieved and modified. Our experience saves you time and cost.
Our specialists have extensive experience of protocol execution. They all operate to a standard set of internal SPGL SOPs and use common test equipment.
Clients may either elect to use our SOPs or may wish to use their own. We can undertake all execution work or train your own operators and supervise execution activities. The choice is yours.
Summary reports are prepared for all protocols executed. These are intended to provide a concise summary document to assist in presenting validation data to regulatory Investigators and assessors. Summary reports are used to present a summary of the data obtained, to interpret the results and to conclude whether the objectives of the study were met. They may contain recommendations for additional process controls or changes to operating procedures. They may also contain recommendations for calibration, re-validation, additional training and change control
Our group contains a number of consultants and specialists with manufacturing, QA and development experience working in major pharmaceutical manufacturing companies. Using this experience, we have been able to assist clients in developing validation plans and rationales for cleaning validation projects.
Our consultants have particular expertise in the field of aseptic processing, antibiotic processing, antibiotic powders, fermentation and biotechnology processes, tablets and capsules and highly automated processes. Our portfolio of projects ranges from products in the phases of clinical development, through new product start-ups, to modification and reengineering of existing processes.
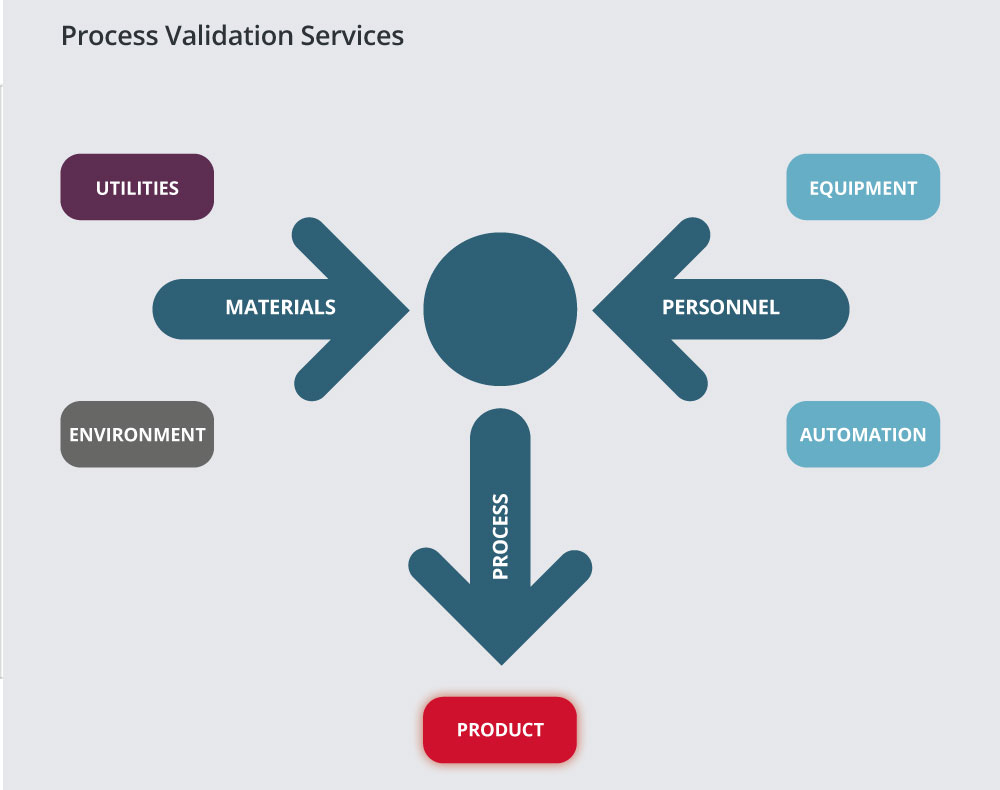
In order to conduct process validation, it is necessary to ensure that all components of the facility validation are complete, i.e. utilities, equipment, environment and automation. The combination of these with the appropriate material and use of trained personnel will result in product which is fit for purpose.
Our experienced engineers and scientists are able to assist clients in developing plans and rationales for process validation projects.
SPGL provides consultancy services on both a technical and regulatory basis for automation and IT projects in a range of market sectors. Our risk-based approach to computer system validation is based on best industry practice and guides such as Good Automated Manufacturing Practice (GAMP). Areas of expertise include:
Manufacturing Systems
IT Systems
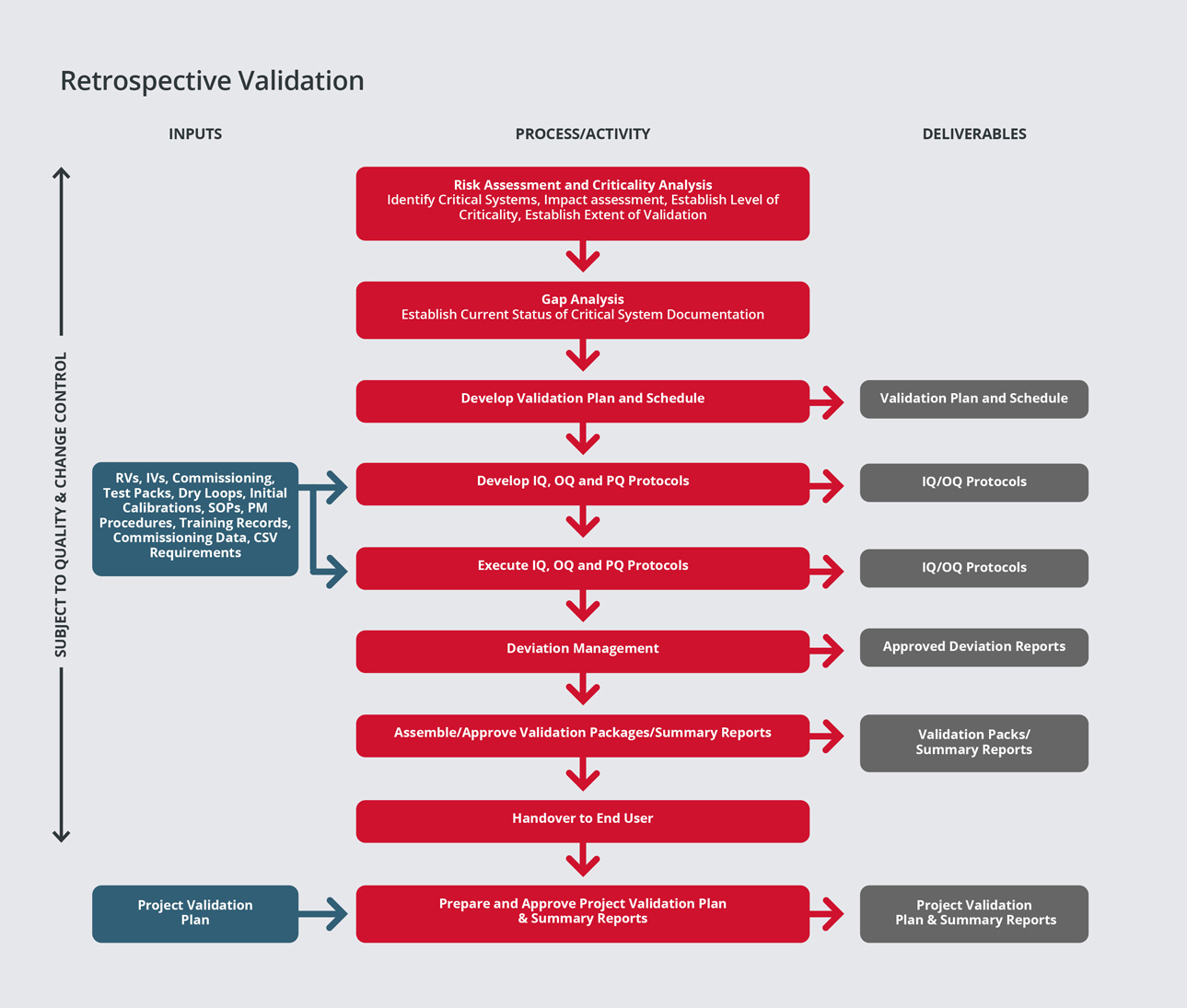
Retrospective validation is used for facilities, processes and process controls in operation use that have not undergone a formally documented validation process. Validation of these facilities, processes and process controls is possible using historical data to provide the necessary documentary evidence that the process is doing what it is believed to do.
At SPGL, we have developed a risk-based approach to retrospective validation, based on best industry practices and guides such as the ISPE.
Our experienced engineers/specialists have extensive retrospective validation experience and are able to assist clients in developing plans, procedures and reports as well as carrying our risk assessments.
SPGL has a leading edge in the industry, having developed a proven methodology for integrating Commissioning & Qualification (C&Q).
We have learned from experience that significant cost and schedule savings can be realized from the integration of engineering, construction, commissioning and qualification activities to address the buildings and all systems (Direct Impact, Indirect Impact, and No Impact) that comprise a pharmaceutical/biotech project.
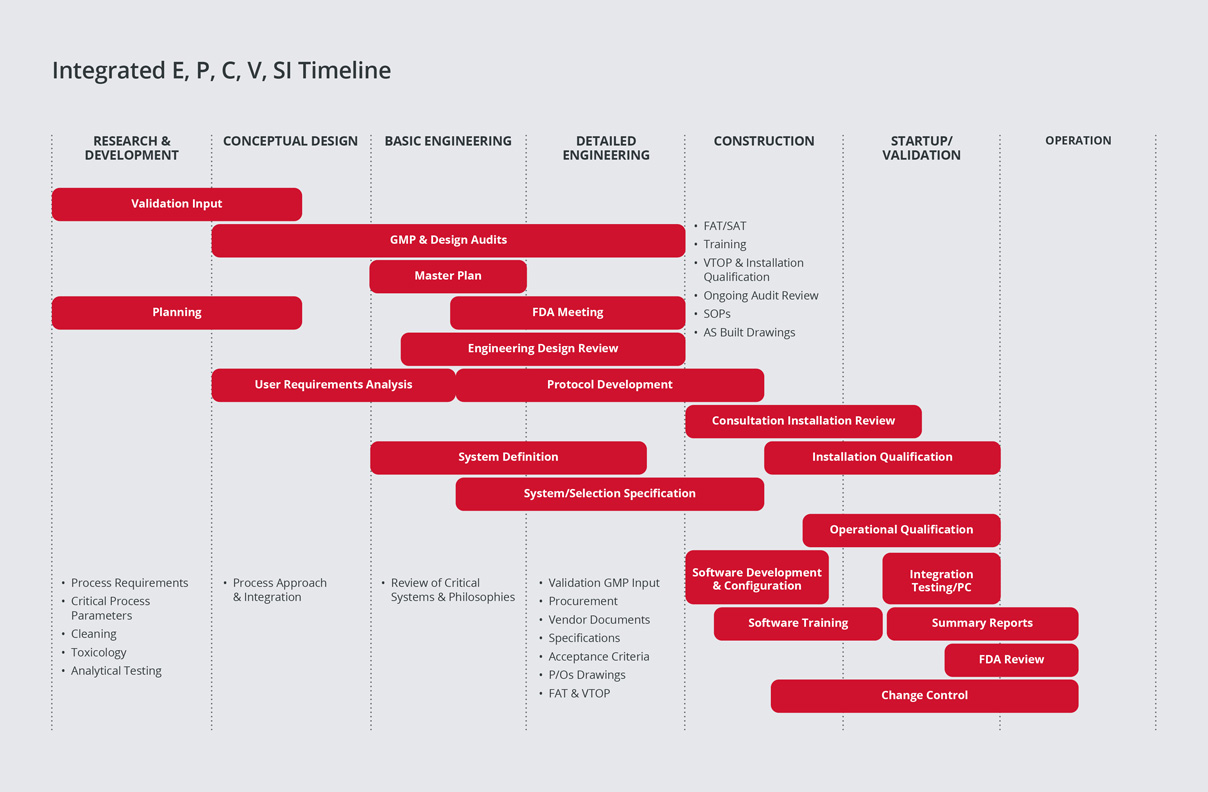
During project execution, our strategy is to closely integrate construction with commissioning and to utilise the project’s enhanced commissioning process to reduce the time taken for qualification, hence reducing ‘Time to Market’.
Our strategy for field quality is to apply regulatory compliance principles to construction activities to ensure a smooth transition into commissioning. Our field quality program ensures proper construction turnover and that systems are ready for commissioning.
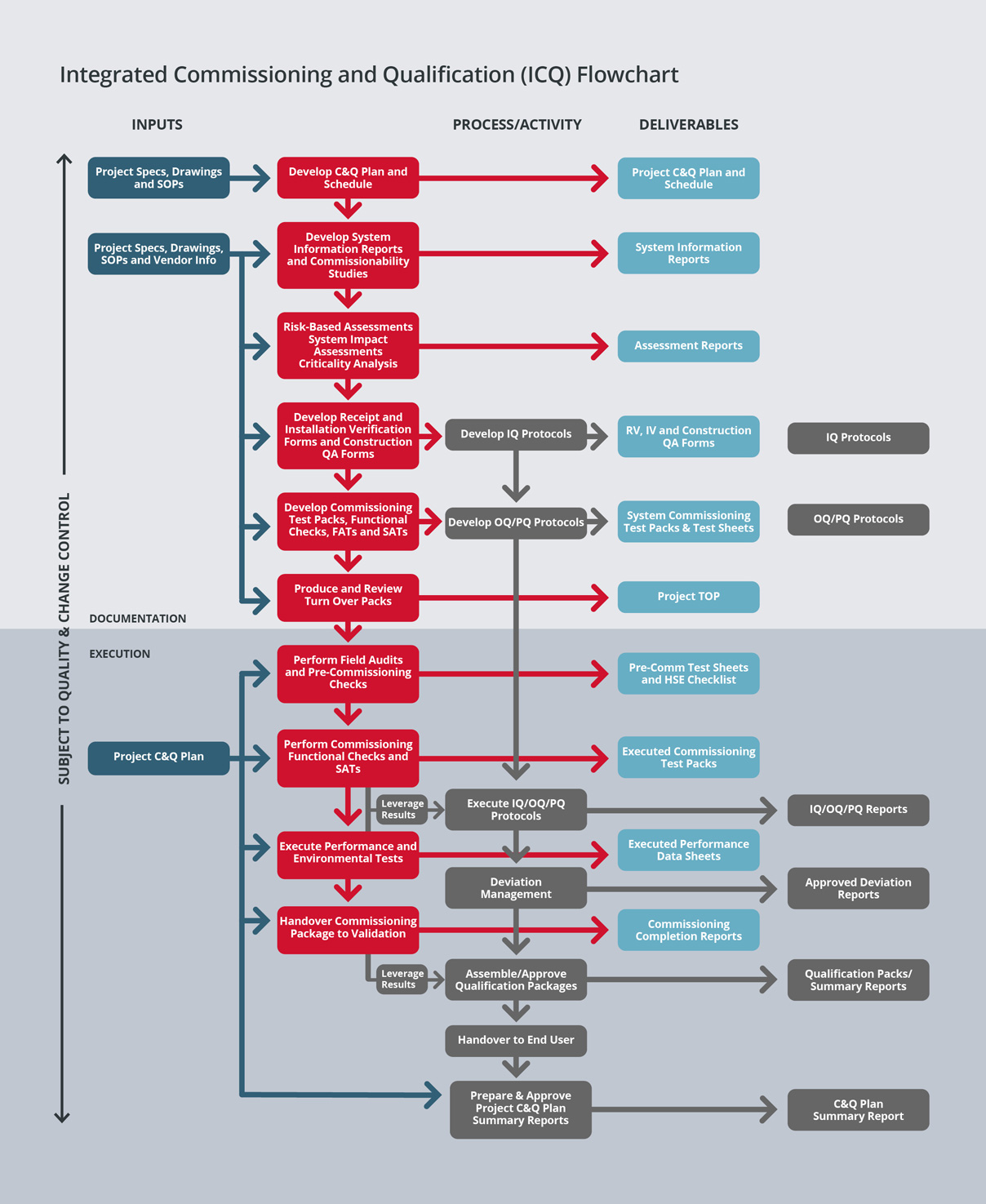
We then perform enhanced commissioning to ensure that commissioning activities and documentation can be leveraged into qualification.
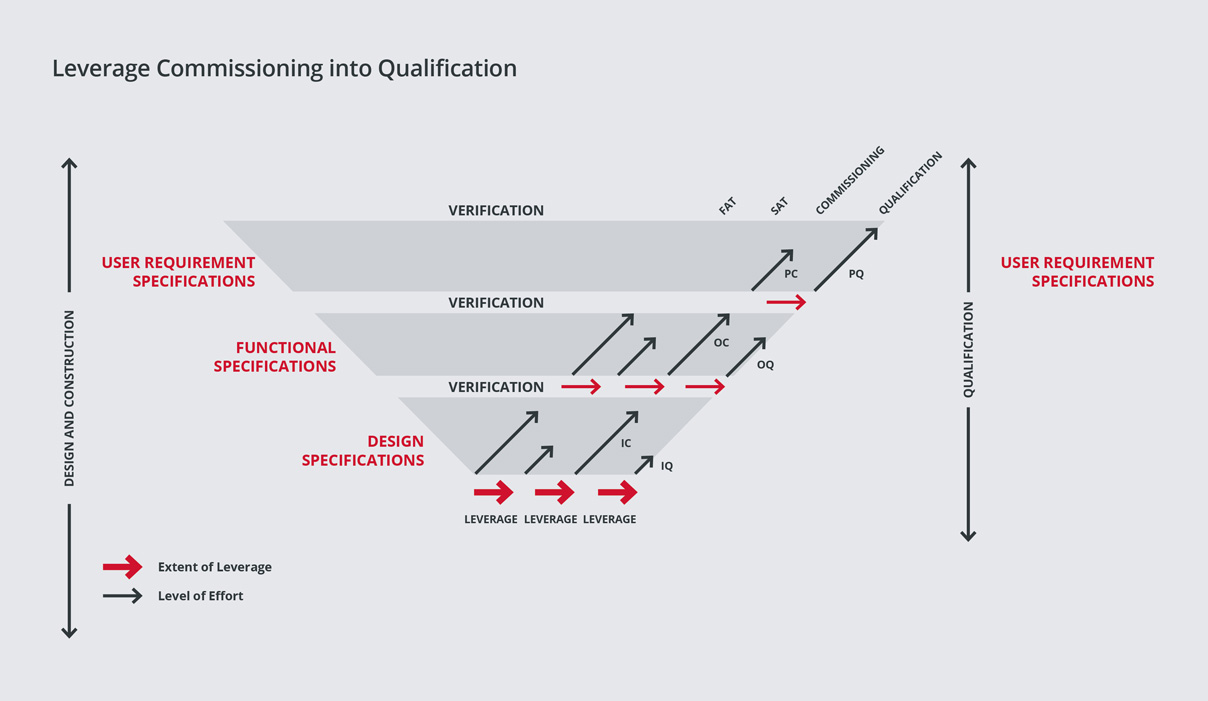
This philosophy is based on the concept that a properly executed and documented commissioning effort will expedite plant delivery. Often the problems that are encountered in qualification are due to incomplete commissioning procedures.
We strongly believe that a fully executed and properly documented commissioning can eliminate many downstream problems and accomplish much of the data gathering required for qualification and plant delivery. By reducing the time for qualification, we lower project cost and save time within the overall project schedule.
Properly planned commissioning should begin during the pre-construction phase of a project or as early as possible. Therefore, we will identify the parameters for commissioning and qualification turnover documents during the pre-construction phase of a project.
Factory Acceptance Test (FAT) plans and Site Acceptance Test (SAT) plans will be developed for pre-purchased equipment and systems. Our goal will be to have the commissioning and closeout documentation requirements.
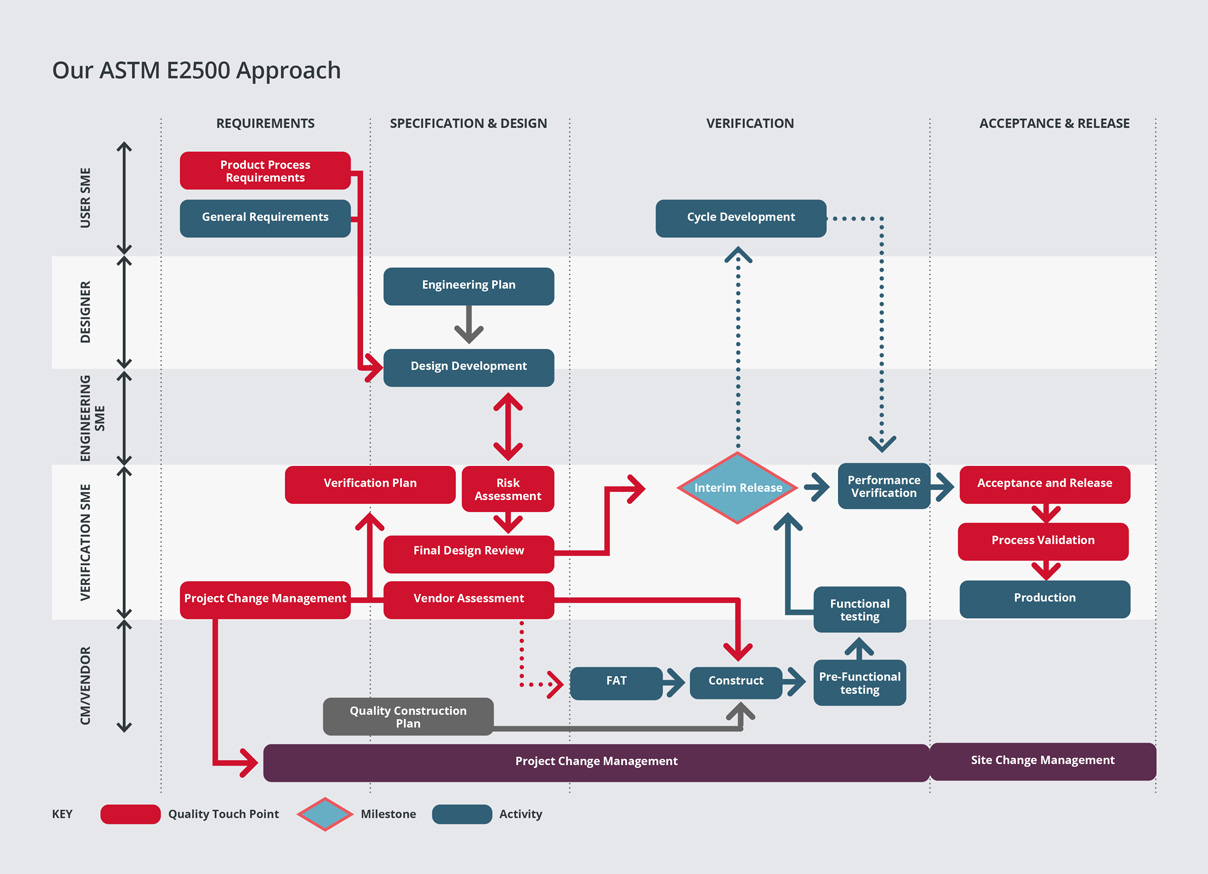
Our Verification Approach is based on the ASTM E2500 standard and it applies the concepts and principles introduced in the FDA initiative, Pharmaceutical cGMPs for the 21st century. It’s a ‘Science and Risk-Based Approach’ that focusses on the ‘critical aspects of manufacturing systems’.
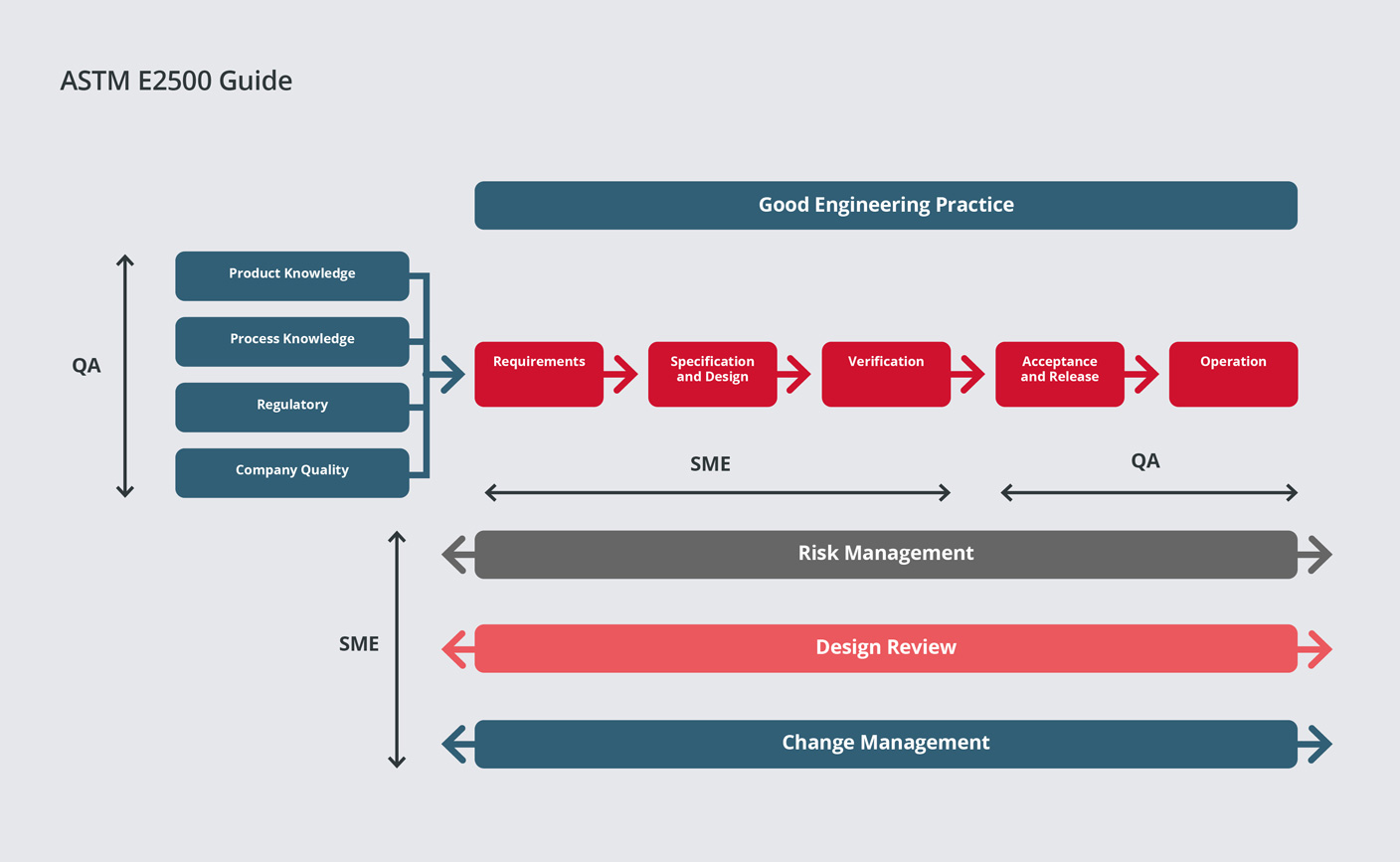
By applying these principles from the design phase there has been a paradigm shift from qualification/validation to ‘quality by design’.
GEP (Good Engineering Practice) and SME (Subject Matter Expert) knowledge underpin the methodology and the use of vendor documentation is an integral part of the approach. The approach can be applied to small and large projects.